April 22nd/23rd 2021
Requirements
Participants must have access to a desktop or laptop computer with a Mac, Linux, or Windows operating system (not a tablet, Chromebook, etc.).
Before the start of the course you should ensure you are able to connect to ARCHER2 using the Setup instructions provided.
You are also required to abide by the ARCHER2 Training Code of Conduct.
Description
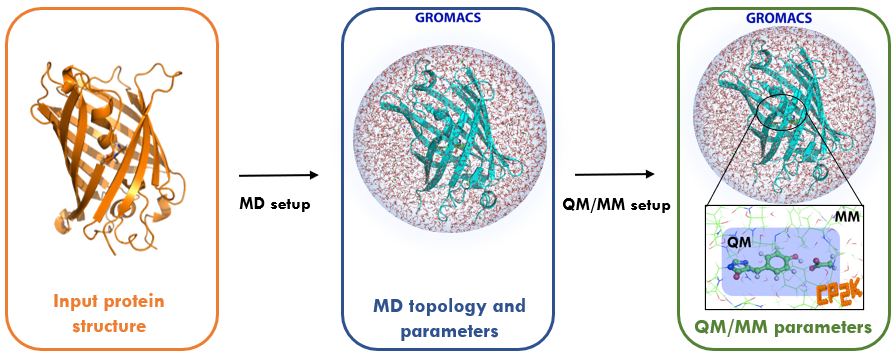
Most classical molecular mechanics (MM) force fields are not sufficiently flexible to model processes in which chemical bonds are broken or formed. Instead, some level of quantum mechanical (QM) description is needed to gain detailed mechanistic insights into reaction mechanisms, e.g. the catalytic action of enzymes, and to compute photochemical properties such as absorption and emission spectra of fluorescent proteins. However biochemical systems (including solvent) are too large to be solved in their entirety at ab initio QM level or efficiently even with a high-quality density functional theory (DFT) approximation. To overcome the cost of a QM description on the one hand versus the limitations of a MM treatment on the other hand, methods have been developed that treat a small critical region of interest at QM level while retaining the computationally cheaper MM force field for the majority of the system.
Despite the simplicity of the QM/MM concept, its application is currently restricted to a relatively small group of specialised expert users often using in-house codes. To unlock the power of QM/MM simulations for the wider biomolecular simulation community, the BioExcel Centre of Excellence has developed an interface between the molecular dynamics code GROMACS, which is widely used in the biomolecular simulation community, and the quantum chemistry package CP2K, which supports calculations under periodic boundary conditions with DFT methods.
This course explains key aspects relating to the use of QM/MM for biomolecular simulation with these two codes and equips participants with the ability to start using GROMACS and CP2K for this purpose in their own research. Participants are provided with the opportunity to gain hands-on practical experience running several example calculations on ARCHER2, the UK national supercomputing service.
Learning outcomes
This course equips participants to perform hybrid QM/MM simulations using the molecular dynamics code GROMACS in combination with the quantum chemistry package CP2K through an interface developed by one of the instructors from the BioExcel Centre of Excellence.
Prerequisites
Participants should be familiar with using GROMACS to perform molecular dynamics simulations. A background in computational quantum chemistry is not strictly required since an introduction of key aspects is provided, but previous familiarity is beneficial. You should be familiar with connecting to a remote machine using ssh and will benefit from experience using an HPC facility as you will execute hands-on practicals on the UK supercomputer ARCHER2.
Contact: For questions or more information, email a.proeme@epcc.ed.ac.uk
This course is delivered by/in collaboration with:

